X_nonIMID, y_nonIMID = prep_data(data_all, 'SLE', 'nonIMID', drop_cols = ["Arthritis","Pleurisy","Pericarditis","Nefritis"])dsDNA_nonIMID = X_nonIMID.dsDNA2.values.reshape(-1,1) # only dsDNA from chip as a vectordsDNA_lab_nonIMID = X_nonIMID.dsDNA1.values.reshape(-1,1)X_nonIMID.drop("dsDNA1", axis=1, inplace=True) # drop clinic dsDNA from multivariable data frame
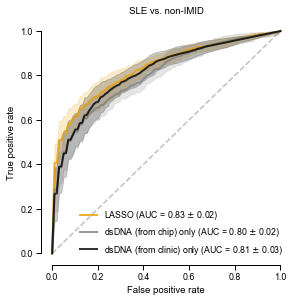
Interestingly, AUC for dsDNA only is around the same value as for the vs. blood bank controls, though this is a much harder classification problem. So dsDNA seems to be the most relevant in this set. Also, it’s barely lower than the XGBoost model…
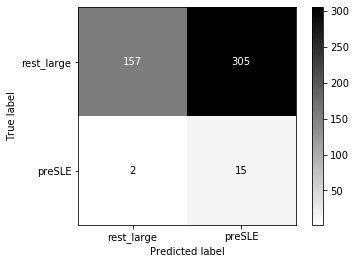
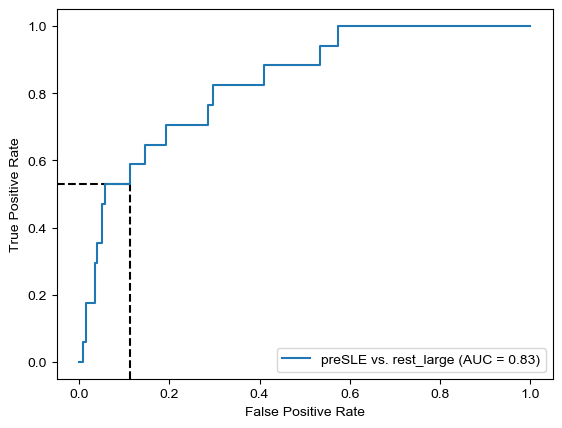
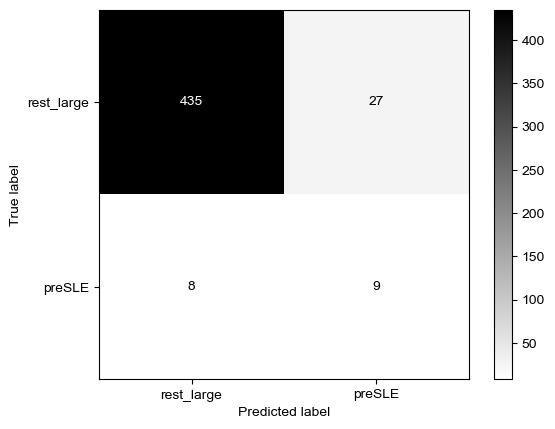
Threshold for classification: 0.5
precision recall f1-score support
rest_large 0.99 0.34 0.51 462
preSLE 0.05 0.88 0.09 17
accuracy 0.36 479
macro avg 0.52 0.61 0.30 479
weighted avg 0.95 0.36 0.49 479
N.B.: "recall" = sensitivity for the group in this row (e.g. preSLE); specificity for the other group (rest_large)
N.B.: "precision" = PPV for the group in this row (e.g. preSLE); NPV for the other group (rest_large)
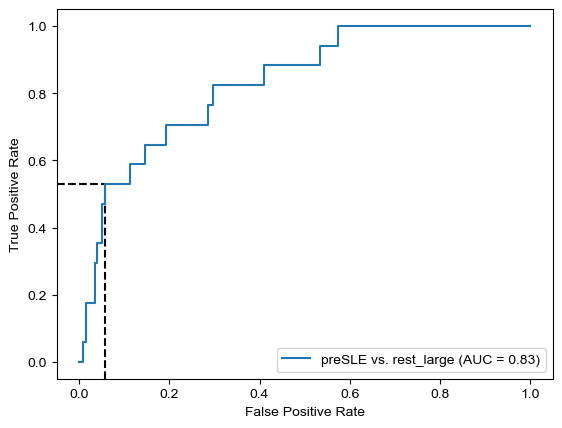
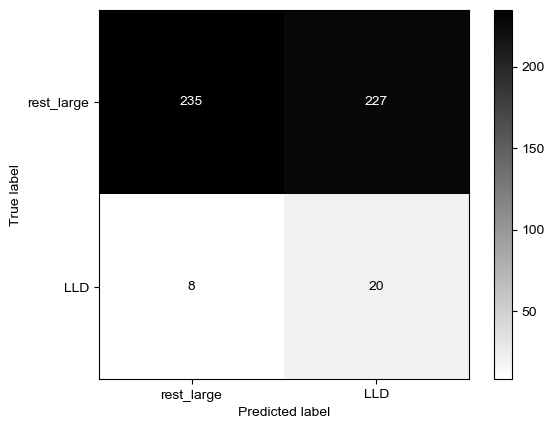
Threshold for classification: 0.5
precision recall f1-score support
rest_large 0.98 0.34 0.50 462
LLD 0.08 0.89 0.14 28
accuracy 0.37 490
macro avg 0.53 0.62 0.32 490
weighted avg 0.93 0.37 0.48 490
N.B.: "recall" = sensitivity for the group in this row (e.g. LLD); specificity for the other group (rest_large)
N.B.: "precision" = PPV for the group in this row (e.g. LLD); NPV for the other group (rest_large)
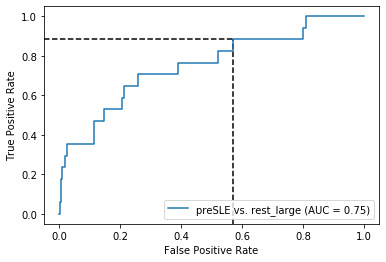
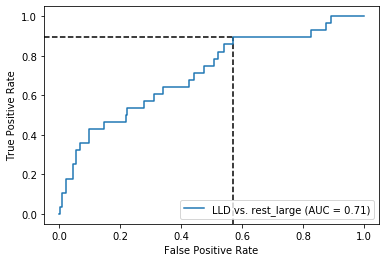
Both for preSLE and LLD, sensitivity is great when only using dsDNA… But specificity is pretty bad
Threshold for classification: 0.5
precision recall f1-score support
BBD 0.38 0.13 0.20 361
IMID 0.46 0.77 0.58 346
accuracy 0.45 707
macro avg 0.42 0.45 0.39 707
weighted avg 0.42 0.45 0.38 707
N.B.: "recall" = sensitivity for the group in this row (e.g. IMID); specificity for the other group (BBD)
N.B.: "precision" = PPV for the group in this row (e.g. IMID); NPV for the other group (BBD)
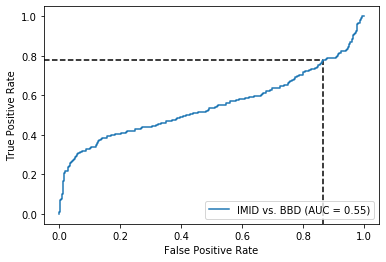
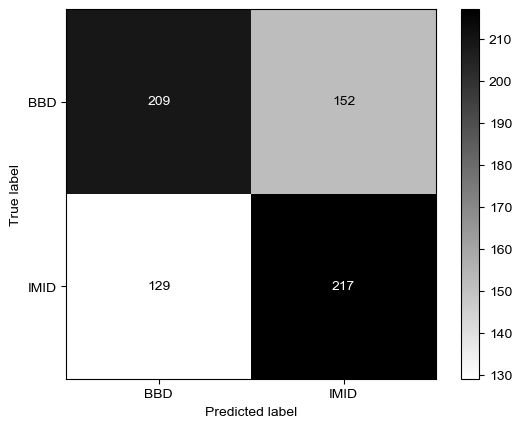
Does not do so well even on healthy controls, so this model is just very sensitive to everything it seems
Logistic regression: Only dsDNA from clinic
clf = LogisticRegression(penalty ='none', max_iter =10000) # increase iterations for solver to converge
Threshold for classification: 0.5
precision recall f1-score support
rest_large 0.00 0.00 0.00 462
preSLE 0.04 1.00 0.07 17
accuracy 0.04 479
macro avg 0.02 0.50 0.03 479
weighted avg 0.00 0.04 0.00 479
N.B.: "recall" = sensitivity for the group in this row (e.g. preSLE); specificity for the other group (rest_large)
N.B.: "precision" = PPV for the group in this row (e.g. preSLE); NPV for the other group (rest_large)
/home/lcreteig/miniconda3/envs/SLE/lib/python3.7/site-packages/sklearn/metrics/_classification.py:1221: UndefinedMetricWarning: Precision and F-score are ill-defined and being set to 0.0 in labels with no predicted samples. Use `zero_division` parameter to control this behavior.
_warn_prf(average, modifier, msg_start, len(result))
Threshold for classification: 0.5
precision recall f1-score support
rest_large 0.00 0.00 0.00 462
LLD 0.06 1.00 0.11 28
accuracy 0.06 490
macro avg 0.03 0.50 0.05 490
weighted avg 0.00 0.06 0.01 490
N.B.: "recall" = sensitivity for the group in this row (e.g. LLD); specificity for the other group (rest_large)
N.B.: "precision" = PPV for the group in this row (e.g. LLD); NPV for the other group (rest_large)
/home/lcreteig/miniconda3/envs/SLE/lib/python3.7/site-packages/sklearn/metrics/_classification.py:1221: UndefinedMetricWarning: Precision and F-score are ill-defined and being set to 0.0 in labels with no predicted samples. Use `zero_division` parameter to control this behavior.
_warn_prf(average, modifier, msg_start, len(result))
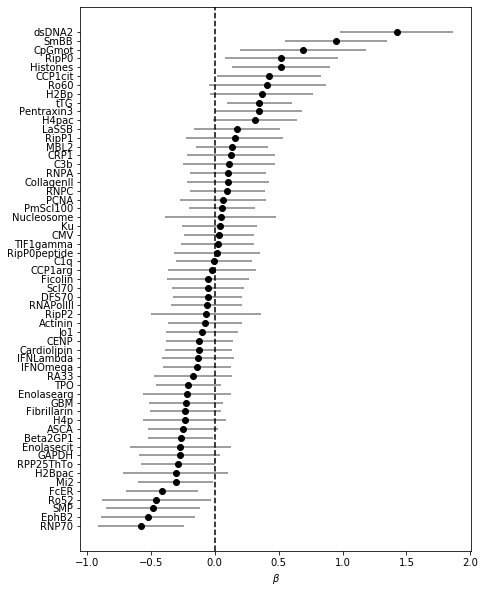
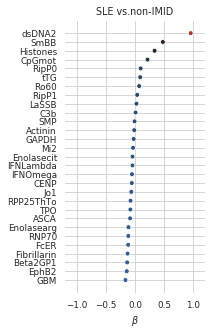
In contrast to blood bank controls, there’s no quasi-complete separation in this model. CIs are wider and less predictors are significant. dsDNA and anti-Smith lead, which is to be expected. CpGmot also significant here. Ro60 quite a bit lower in the rank, and anti-correlated to Ro52…
(<matplotlib.axes._subplots.AxesSubplot at 0x7f7c207dc690>,
<matplotlib.axes._subplots.AxesSubplot at 0x7f7c2292dc10>,
<matplotlib.axes._subplots.AxesSubplot at 0x7f7c20789610>)
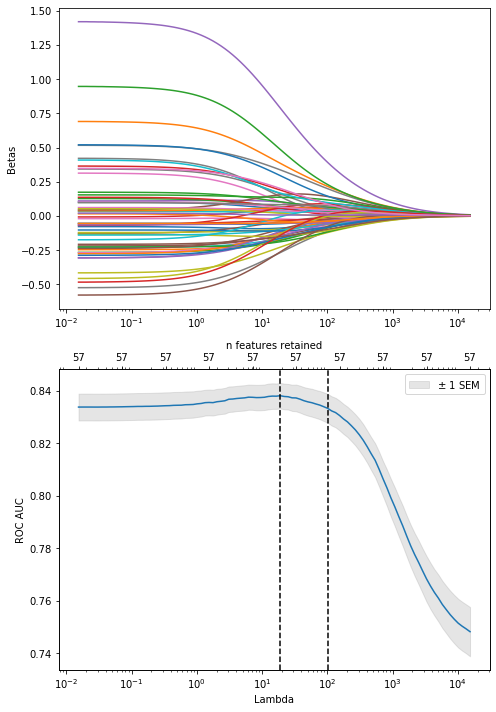
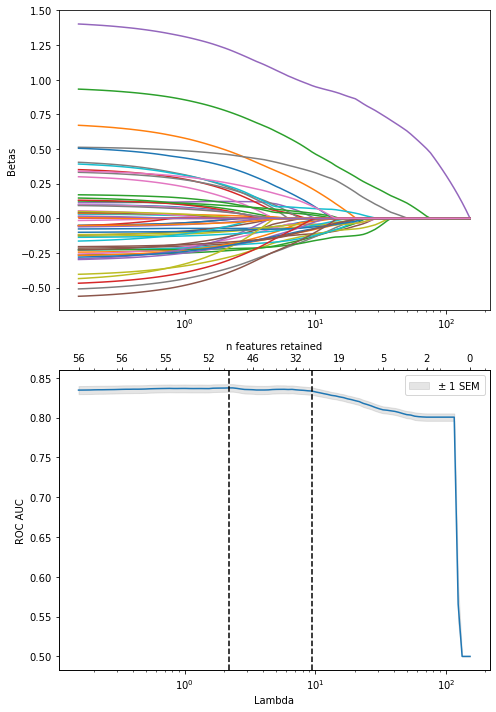
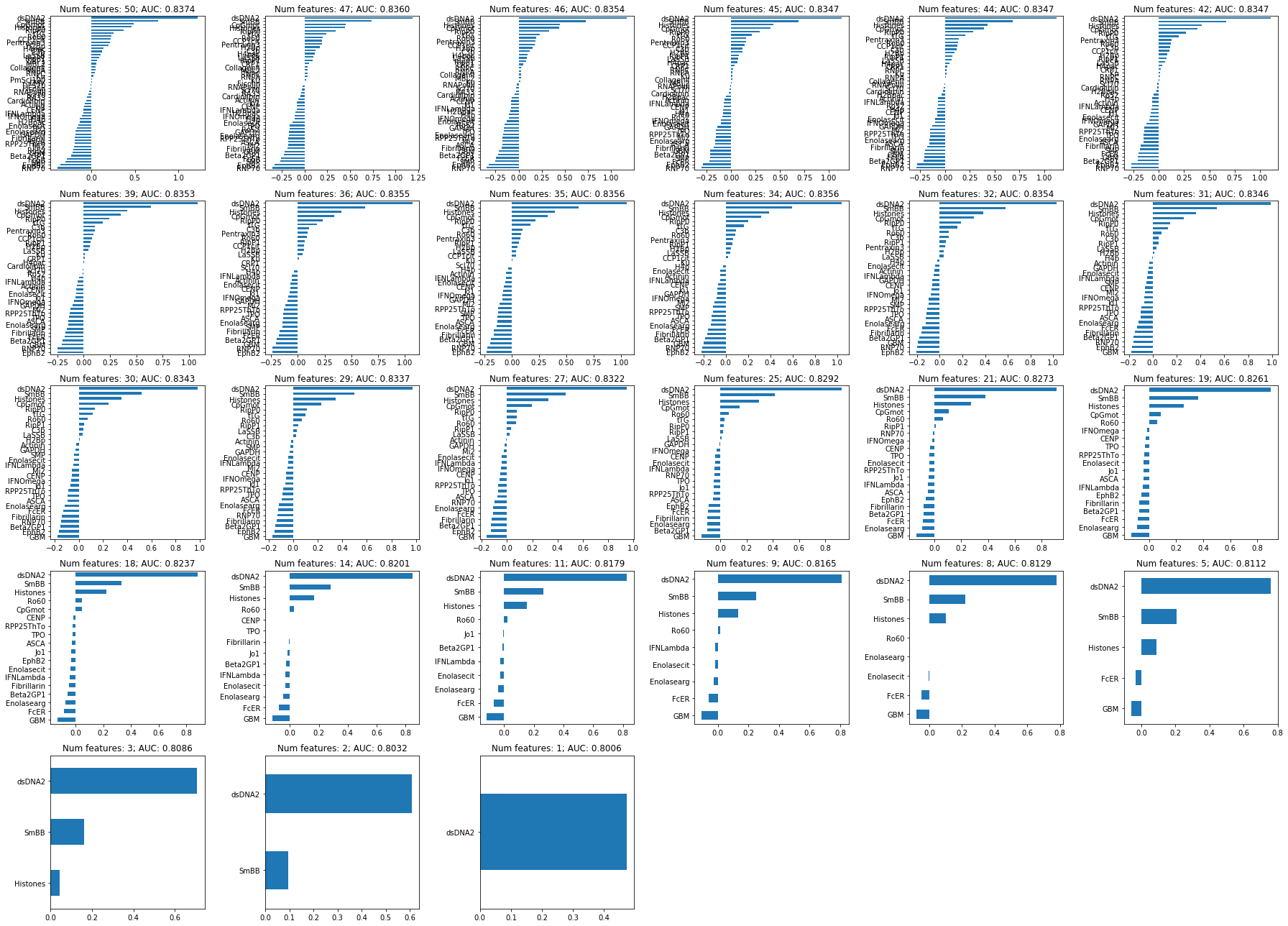
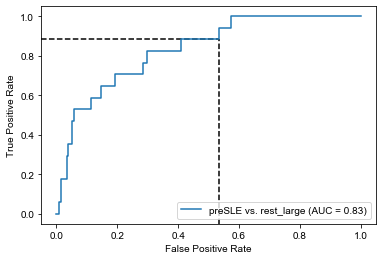
So again, dsDNA is really the dominant feature here. But, to maximize performance, it might be best to regularize until about half of the original features are included, as performance starts to decline monotonically from that point forwards
with sns.plotting_context("paper"): plt.rcParams["font.family"] ="Arial" plt.rcParams['pdf.fonttype'] =42# this causes the pdf to be editable, but it also embeds the font, significantly increasing the size fig, ax = plt.subplots(figsize=(4.5, 4.5)) tprs, aucs = calc_roc_cv(lasso_nonIMID.best_estimator_,cv,Xp1_nonIMID,y_nonIMID) fig, ax = plot_roc_cv(tprs, aucs, fig, ax, fig_title='SLE vs. non-IMID', line_color='#E69F00', legend_label='LASSO') tprs, aucs = calc_roc_cv(lr_dsDNA_nonIMID,cv,dsDNA_nonIMID,y_nonIMID) fig, ax = plot_roc_cv(tprs, aucs, fig, ax, reuse=True, line_color='gray', legend_label='dsDNA (from chip) only') tprs, aucs = calc_roc_cv(lr_dsDNA_lab_nonIMID,cv,dsDNA_lab_nonIMID,y_nonIMID) fig, ax = plot_roc_cv(tprs, aucs, fig, ax, reuse=True, line_color='black', legend_label='dsDNA (from clinic) only') sns.despine(fig=fig,ax=ax, trim=True) plt.legend(frameon=False) plt.show() fig.savefig('roc_SLE_nonIMID.png', bbox_inches='tight', dpi=300, transparent=True) fig.savefig('roc_SLE_nonIMID.pdf', bbox_inches='tight', transparent=True)
Non-zero coefficients of the model selected with the 1SE rule:
coefs_final_nonimid = (pd.Series(lasso_nonIMID.best_estimator_.named_steps.clf.coef_.squeeze(), index = X_nonIMID.columns)[lambda x: x!=0].sort_values())coefs_final_nonimid
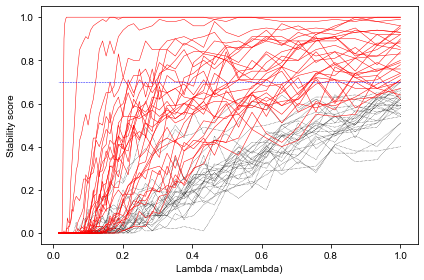
In contrast to the blood bank controls, refitting with the chosen lambda to the whole dataset here seems to include much more features than during CV: 29 vs < 20. Might suggest that the included features are less stable. So let’s examine stability selection again:
%%timeselector_nonIMID = StabilitySelection(base_estimator=pipe, lambda_name='clf__C', lambda_grid=Cs_lasso_nonIMID[np.argmax(lasso_nonIMID.cv_results_["mean_test_score"]):], random_state=40) # range from lambda with highest score to lambda_maxselector_nonIMID.fit(Xp1_nonIMID, y_nonIMID)
CPU times: user 9min 4s, sys: 0 ns, total: 9min 4s
Wall time: 9min 7s
Many of these seem to be selected as well by the 1SE LASSO model, but not all. For instance, the two variables with the lowest (positive) coefficients (RipP1, LaSSB) are missing here. At least in the case of RipP1, that might be because it’s highly correlated with other variables. When LASSO selects one of these, RipP1 will not be, driving down its inclusion probability.
All variables with a stability score of > .80 are included in the 1SE LASSO model, but below there are some differences
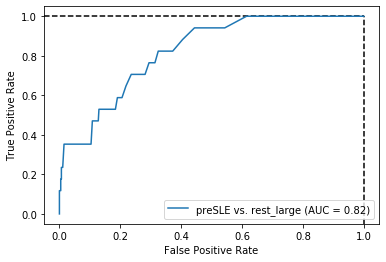
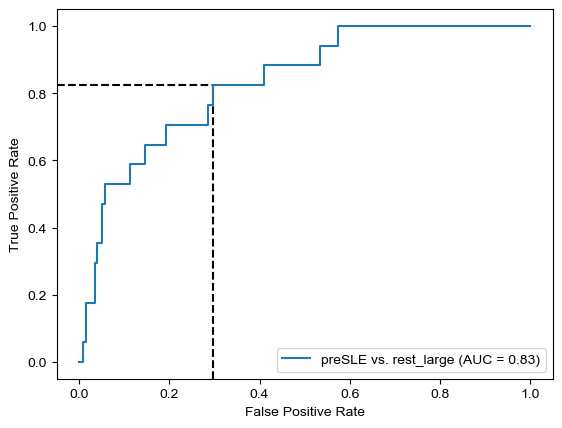
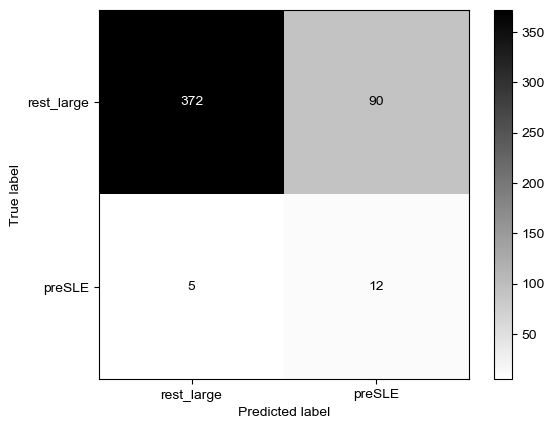
Threshold for classification: 0.5
precision recall f1-score support
rest_large 0.99 0.51 0.67 462
preSLE 0.06 0.88 0.12 17
accuracy 0.52 479
macro avg 0.53 0.70 0.39 479
weighted avg 0.96 0.52 0.65 479
N.B.: "recall" = sensitivity for the group in this row (e.g. preSLE); specificity for the other group (rest_large)
N.B.: "precision" = PPV for the group in this row (e.g. preSLE); NPV for the other group (rest_large)
Sensitivity is the same as when only using dsDNA, but specificity did improve quite a bit
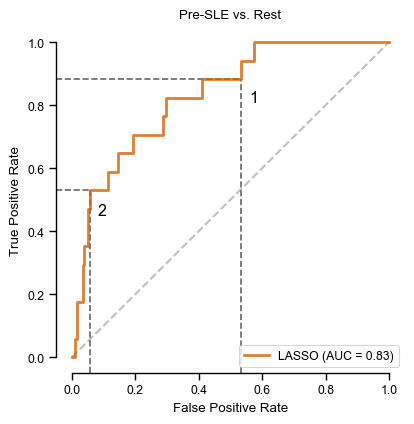
Figure 3:
X_test, y_test = prep_data(X_test_df, 'preSLE', 'rest_large', ["Arthritis","Pleurisy","Pericarditis","Nefritis"] + ["dsDNA1"])# ROC curvethreshold1=0.5threshold2=0.84fpr, tpr, thresholds = roc_curve(y_test, lasso_nonIMID.predict_proba(X_test+1)[:,1])thr_idx1 = (np.abs(thresholds - threshold1)).argmin() # find index of value closest to chosen thresholdthr_idx2 = (np.abs(thresholds - threshold2)).argmin() # find index of value closest to chosen thresholdsns.reset_defaults()with sns.plotting_context("paper"): plt.rcParams["font.family"] ="Arial" plt.rcParams['pdf.fonttype'] =42# this causes the pdf to be editable, but it also embeds the font, significantly increasing the size fig, ax = plt.subplots(figsize=(4.5, 4.5)) ax.plot([0, 1], [0, 1], linestyle='--', lw=1.5, color='k', alpha=.25) ax.set(title="Pre-SLE vs. Rest", xlim=[-0.05, 1.05], xlabel='False positive rate', ylim=[-0.05, 1.05], ylabel='True positive rate') ymin, ymax = ax.get_ylim(); xmin, xmax = ax.get_xlim() plt.vlines(x=fpr[thr_idx1], ymin=ymin, ymax=tpr[thr_idx1], color='k', alpha=.6, linestyle='--', axes=ax) # plot line for fpr at threshold plt.hlines(y=tpr[thr_idx1], xmin=xmin, xmax=fpr[thr_idx1], color='k', alpha=.6, linestyle='--', axes=ax) # plot line for tpr at threshold plt.vlines(x=fpr[thr_idx2], ymin=ymin, ymax=tpr[thr_idx2], color='k', alpha=.6, linestyle='--', axes=ax) # plot line for fpr at threshold plt.hlines(y=tpr[thr_idx2], xmin=xmin, xmax=fpr[thr_idx2], color='k', alpha=.6, linestyle='--', axes=ax) # plot line for tpr at threshold plot_roc_curve(lasso_nonIMID, X_test+1, y_test, name ="LASSO", ax=ax, color='#D55E00', lw=2, alpha=.8) plt.text(0.56,0.81, '1', fontsize='large') plt.text(0.08,0.45, '2', fontsize='large') sns.despine(fig=fig,ax=ax, trim=True) fig.savefig('roc_pre-SLE_Rest.png', bbox_inches='tight', dpi=300, transparent=True) fig.savefig('roc_pre-SLE_Rest.pdf', bbox_inches='tight', transparent=True)
Threshold for classification: 0.6
precision recall f1-score support
rest_large 0.99 0.67 0.80 462
preSLE 0.08 0.82 0.15 17
accuracy 0.67 479
macro avg 0.54 0.75 0.48 479
weighted avg 0.96 0.67 0.78 479
N.B.: "recall" = sensitivity for the group in this row (e.g. preSLE); specificity for the other group (rest_large)
N.B.: "precision" = PPV for the group in this row (e.g. preSLE); NPV for the other group (rest_large)
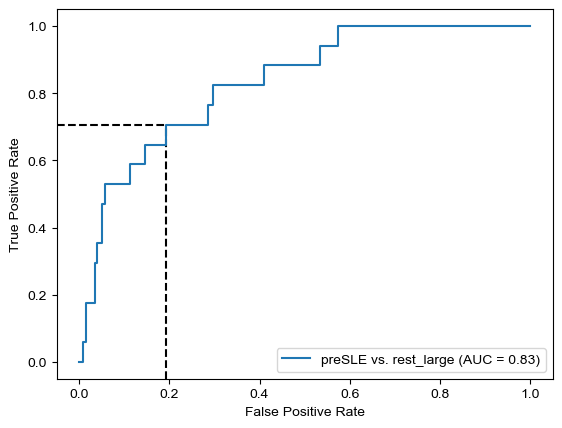
Threshold for classification: 0.7
precision recall f1-score support
rest_large 0.99 0.81 0.89 462
preSLE 0.12 0.71 0.20 17
accuracy 0.80 479
macro avg 0.55 0.76 0.54 479
weighted avg 0.96 0.80 0.86 479
N.B.: "recall" = sensitivity for the group in this row (e.g. preSLE); specificity for the other group (rest_large)
N.B.: "precision" = PPV for the group in this row (e.g. preSLE); NPV for the other group (rest_large)
Threshold for classification: 0.8
precision recall f1-score support
rest_large 0.98 0.91 0.95 462
preSLE 0.18 0.53 0.27 17
accuracy 0.90 479
macro avg 0.58 0.72 0.61 479
weighted avg 0.95 0.90 0.92 479
N.B.: "recall" = sensitivity for the group in this row (e.g. preSLE); specificity for the other group (rest_large)
N.B.: "precision" = PPV for the group in this row (e.g. preSLE); NPV for the other group (rest_large)
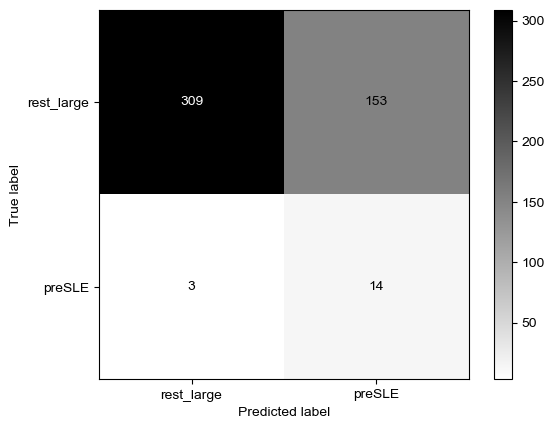
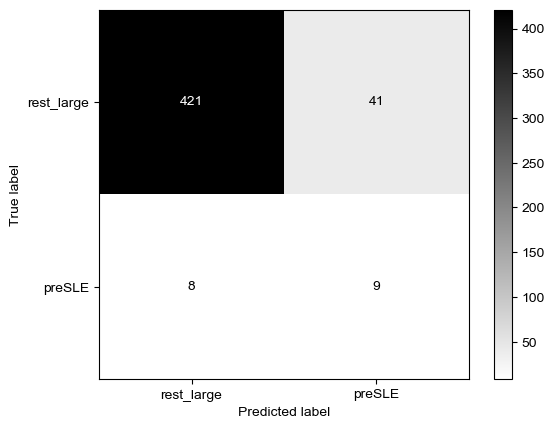
Threshold for classification: 0.84
precision recall f1-score support
rest_large 0.98 0.94 0.96 462
preSLE 0.25 0.53 0.34 17
accuracy 0.93 479
macro avg 0.62 0.74 0.65 479
weighted avg 0.96 0.93 0.94 479
N.B.: "recall" = sensitivity for the group in this row (e.g. preSLE); specificity for the other group (rest_large)
N.B.: "precision" = PPV for the group in this row (e.g. preSLE); NPV for the other group (rest_large)
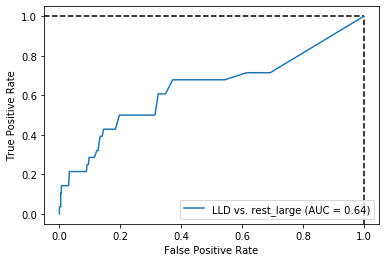
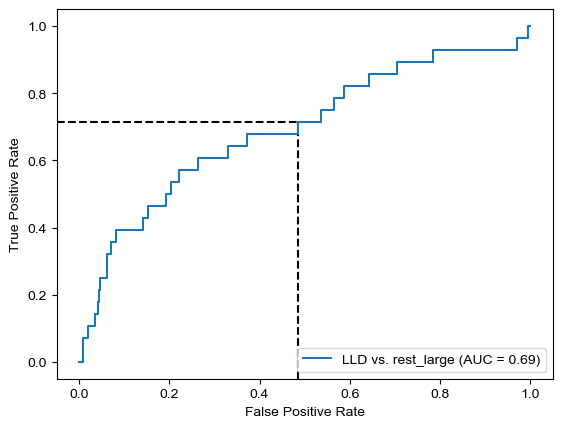
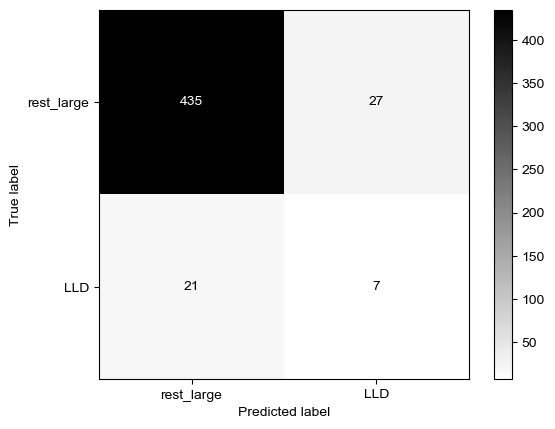
Threshold for classification: 0.5
precision recall f1-score support
rest_large 0.97 0.51 0.67 462
LLD 0.08 0.71 0.15 28
accuracy 0.52 490
macro avg 0.52 0.61 0.41 490
weighted avg 0.92 0.52 0.64 490
N.B.: "recall" = sensitivity for the group in this row (e.g. LLD); specificity for the other group (rest_large)
N.B.: "precision" = PPV for the group in this row (e.g. LLD); NPV for the other group (rest_large)
Sensitivity was actually better for dsDNA only… But again, specificity here is much higher
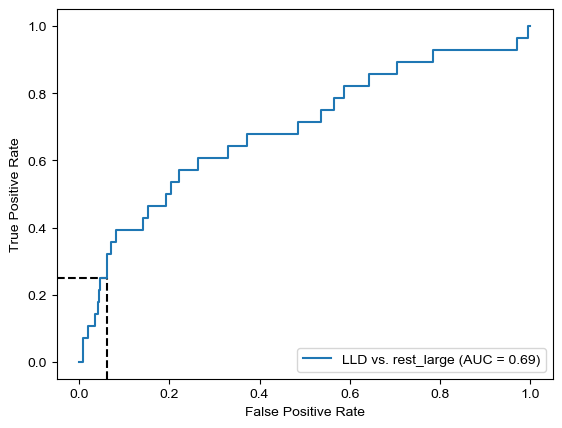
Threshold for classification: 0.84
precision recall f1-score support
rest_large 0.95 0.94 0.95 462
LLD 0.21 0.25 0.23 28
accuracy 0.90 490
macro avg 0.58 0.60 0.59 490
weighted avg 0.91 0.90 0.91 490
N.B.: "recall" = sensitivity for the group in this row (e.g. LLD); specificity for the other group (rest_large)
N.B.: "precision" = PPV for the group in this row (e.g. LLD); NPV for the other group (rest_large)
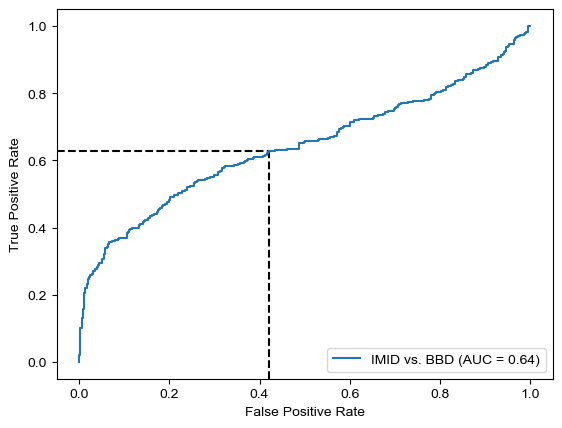
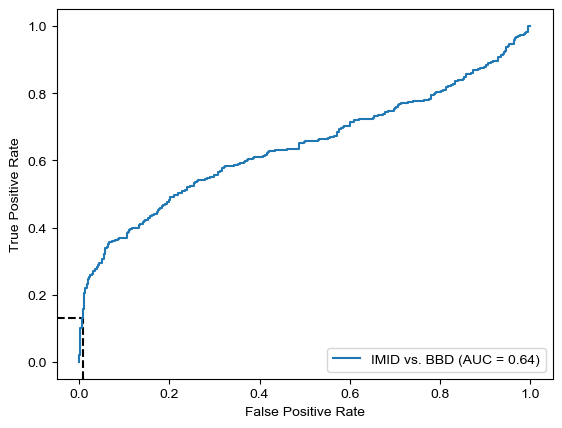
Threshold for classification: 0.5
precision recall f1-score support
BBD 0.62 0.58 0.60 361
IMID 0.59 0.63 0.61 346
accuracy 0.60 707
macro avg 0.60 0.60 0.60 707
weighted avg 0.60 0.60 0.60 707
N.B.: "recall" = sensitivity for the group in this row (e.g. IMID); specificity for the other group (BBD)
N.B.: "precision" = PPV for the group in this row (e.g. IMID); NPV for the other group (BBD)
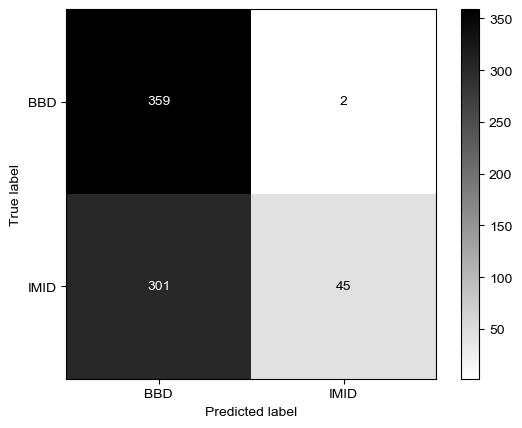
Threshold for classification: 0.84
precision recall f1-score support
BBD 0.54 0.99 0.70 361
IMID 0.96 0.13 0.23 346
accuracy 0.57 707
macro avg 0.75 0.56 0.47 707
weighted avg 0.75 0.57 0.47 707
N.B.: "recall" = sensitivity for the group in this row (e.g. IMID); specificity for the other group (BBD)
N.B.: "precision" = PPV for the group in this row (e.g. IMID); NPV for the other group (BBD)